Results
主な研究成果
チームIでは,長鎖RNAについて二次構造解析を行い,二次構造を形成する可能性の高い領域を検出する方法の開発,および検出した領域に対応するRNA断片の二次構造あるいは立体構造をNMR法によって解析する手法の開発を行った.また,RNA断片と他の分子との相互作用を迅速に解析するための手法の開発を進めた.以下にチームIの成果の概要を示す.
I−1.神経特異的mRNA であるHAR1F RNAの二次構造解析
ほ乳類での保存性が高く,同時に,ヒトにおいて変異が認められる領域のひとつであるHAR1から発現しているnon-coding RNAのHAR1F RNAについて,特定の領域の二次構造がヒトとチンパンジーで異なっていることが報告されている.そこで,このRNAについて,vsfold/GenoPoemics(vsfold/GP)を用いて二次構造解析を行った.その結果,特定の領域に二次構造形成が示された.特に,ヒトとチンパンジーで二次構造が異なることが指摘されている領域で比較的安定な二次構造が形成されることが示された.このように,vsfold/GPを用いることによって,長鎖RNAの二次構造の特徴を効果的に描き出すことができることが示された.ヒトとチンパンジーで異なる二次構造が予測された3つの領域のうち,脊椎動物で保存されているのは,2番目の領域のみであり,生物学的な重要性は,おそらく2番目の領域にあると判断できた.
このように,vsfold/GPによって二次構造の比較が効果的に行えることが示された.今後は,二次構造の差が見られた領域について,実際にNMR法による構造解析を行い,その立体構造の違いについて明らかにしていきたい.
I−2.HCVゲノムRNAにおける構造領域の機能の推定
HCVゲノムが自立的に複製する細胞を用いて複製を抑制する宿主要因を単離するために,HSV チミジンキナーゼ(TK)を利用したスクリーニングの系を作成した際に,TK遺伝子が入っていないレプリコンを持つ細胞では,レプリコンが自立的に複製したのに対して,TK-neoを持つレプリコンは自立複製しなかった.このHCVレプリコンにおけるHCV RNAの自己複製が阻害される現象について,本プロジェクトで開発を進めている手法を適用し,その二次構造を解析した.vsfold/GPによって人為的改変HCV RNAの5'領域の二次構造を解析した結果,いくつかの二次構造が予測された.今後,これらの構造と自己複製阻害との関連について解析する必要がある.なお,HCV RNAのIRES領域にいくつかの二次構造が推定されており,これらはすでに解析されているIRESの二次構造とよく一致していた.このことは本手法の有効性を示すものである.
I−3.免疫機能に関与する長鎖non coding RNAの新規機能の検索
免疫機能の関与する分子のmRNAの3′非翻訳領域(3′-UTR)に着目し,vsfold/GPによって特徴的な構造を見出せるか,検討を行った.
TNFなど炎症に関わる分子のmRNAの3'-UTRには,AUUUA配列を特徴とするAU rich element(ARE)と呼ばれる領域がある.そこで,これらのmRNAについてvsfold/GPで解析したところ,ARE領域の3'側に二次構造形成が予測された.また,サイトカインのような分泌タンパク質やtoll-like receptor(TLR)などのmRNAについても,AREに近接してループ構造の形成が予測された.これらの予測された二次構造について,比較検討を行っている.また,mRNA編集酵素であるAPOBEC1が,IL8のmRNAの3'-UTRのAREと相互作用することが最近見出された.そこで, APOBEC1の認識に必要な構造要素の解明をめざし,vsfold/GPによってIL8 mRNAの二次構造の解析を行った.その結果,AREが集中している領域において,安定ではないが二次構造の形成が予測された.これらの結果は,vsfold/GPが様々なmRNAの構造領域を抽出できることを示している.
I−4.翻訳エンハンサー活性を有するOsMac遺伝子群5'-UTRの構造解析
東京理科大学の島田浩章博士との共同研究として,植物由来mRNAを対象とした構造解析を進めた.イネのOsMac遺伝子群のmRNAは長い5'-UTRをもち,これが下流のopen reading frame(ORF)の翻訳量を10〜20倍に促進することが発見されている.そこで,vsfold/GPによってOsMac1 およびOsMac3の5'-UTRの二次構造解析を行った.
OsMac1遺伝子からは,選択的スプライシングによって3種類のmRNAが合成されるが,このうち5'UTRがもっとも長いmRNAが効率良く翻訳されることが知られており,この配列について二次構造予測を行った.その結果,いくつかの安定なヘアピンが予測されたが,機能部位には安定なステムループは予測されなかった.一方,OsMac3の5'-UTRについてもvsfold/GPによる解析を行った結果,2つの比較的安定な二次構造が予測された.すでにOsMac3 の5'UTRに由来するRNA断片についてのNMR解析をスタートさせており,今後,これらの二次構造あるいは立体構造を比較することによって,翻訳促進に関与する機能構造を明らかにしたいと考えている.
I−5.LINE RNAと逆転写酵素の認識特異性の決定要因
散在性核内反復配列(LINE)の逆転写酵素認識部位と考えられているRNA領域について,逆転写酵素認識メカニズムを明らかにすることを目的とし,東京工業大学の梶川正樹博士らとの共同研究として立体構造解析を進めた.
既にウナギのLINE RNA(UnaL2,17残基)の逆転写酵素結合部位のステムループの立体構造を決定しており,逆転写酵素の認識においてG8が重要であり,U10がその構造を支えていることを示した.一方,ゼブラフィッシュにはUnaL2と極めて類似したZfL2-2と,少し差異のあるZfL2-1がある.塩基配列の比較から, ZfL2-1では,UnaL2/ZfL2-2におけるU10の位置にステムループが挿入されており,G8に対応する位置はA10となっている.
ZfL2-1のステムループの立体構造を決定したところ,A10は,UnaL2/ZfL2-2の場合と同様に5'側とスタッキングしており,ループの内側を向いていた(PDB ID: 2RVO,BMRB ID: 11607).ZfL2-1においてもA10が逆転写酵素による認識特異性に関与しているかどうかを調べるため,この残基をG残基に変えたRNA(ZfL2-1-G10)を調製し,NMRスペクトルの解析を行った.その結果,基本的な構造はZfL2-1と同じであることが分かった.
共同研究者の梶川博士らによって,ゼブラフィッシュLINEの逆転写酵素においてRNAの特異的な認識に関与している領域が明らかにされた.そこで,その領域に対応するペプチドとUnaL2/ZfL2-2およびZfL2-1との相互作用解析を行った.ペプチドについては,NMR解析用の安定同位体標識された試料を調製することが必要なため,無細胞タンパク質合成系を利用した新しい方法を開発した.これは,安定同位体標識化合物のメーカーである大陽日酸株式会社と共同で,理化学研究所NMR施設の産学連携無償利用課題として実施した(外部利用課題13-700-007, 14-700-012).このペプチドを用いて,RNAとの相互作用をゲルシフト法およびNMR法において解析したところ, 逆転写酵素によるZfL2-1およびZfl2-2の識別にG8およびA10が重要であることが明らかとなった.
I−6.HCVゲノムRNAにおける構造領域の立体構造解析
C型肝炎ウイルス(HCV)のゲノムRNAにおいて,いくつかの分子内相互作用が,その複製や翻訳に重要であることが示されている.本プロジェクトでは,NS5Bタンパク質の遺伝子領域に存在するステムループである5B SL3.2と3'-UTRのX regionに存在するX-tail SL2との相互作用(相互作用A),および,5Bのステムループとその上流との相互作用(相互作用B)に着目し,NMR法による解析を進めた
(相互作用A)5B SL3.2とX-tail SL2に由来するRNAについてのNMRスペクトルの解析から,複合体形成によってそれぞれのステムループの構造は大きくは変化しないことが分かった.つぎに複合体について立体構造解析を進めが,キッシングループ周辺の拘束条件が少ないこともあり,ステムループ間の位置関係は決定できなかった.そこで,RDCに由来する大局的な構造情報(各塩基の向きの情報)を導入して構造計算を行ったところ,ステムループ間の位置関係が限定された.このように,RDC情報がRNAの立体構造解析において重要であることが示された.(相互作用B)5Bのステムループとその上流に由来する2つのRNAについて,それぞれ単体のNMRスペクトルと複合体のNMRスペクトルを比較したところ,イミノプロトンシグナルに変化が観測された.これらは,複合体形成による構造変化および分子間塩基対の形成を反映していると考えている.5Bのステムループのシグナルについては変化していないものもあることから,推定どおり,バルジループで相互作用していると考えられる.現在,複合体の立体構造の決定をめざし,NMRシグナルの解析を進めている.
I−7.NMR法による長鎖RNAの構造解析手法の開発
これまでのRNAの立体構造解析の多くは,機能性RNAの一部のヘアピンを切り出して行われており,実際に機能を果たすRNAの部分構造であることが多い.このため,これまでは,部分構造を組み合わせることで全体の機能を類推する研究が多数であった.そこで,特定の立体構造を形成する100ntから300nt程度の長さの機能性RNAの立体構造を NMR法によって解析し,機能と構造の相関を解明する手法を開発することを目的とした. (1)HCV IRESの解析: 100塩基程度の機能性RNAのモデルとして,まずHCV由来のIRESを選んだ.IRESのコア領域に相当する91塩基のRNAについて,全長と2種の断片を調製し,そのNMRスペクトルを測定した.その結果,全長のスペクトルは,各断片のイミノプロトンシグナルの重ね合わせとほぼ一致した.このことから,IRESのコア領域は推定した二次構造を形成していることが示唆された.また,コア領域はその活性にMg2+が必須である. NMR法によるMg2+の添加実験を行った結果,コア領域は活性型(Mg2+結合型)と不活性型で立体構造が異なることが示唆された. (2)高度好熱菌由来リボスイッチの解析: 次に当研究室で発見したThermus thermophilus のTPPリボスイッチを解析対象とした.vsfold5で予測された二次構造に基づき,91残基のRNA(TtTPP91)をデザインし,NMR法によってTPPによる滴定実験を行った結果,TtTPP91とTPPは1対1で複合体を形成していることが示唆された.また,TPP非存在下と結合時のNMRスペクトルを比較しすることによって,TPPの結合によって,複数の新たな水素結合が形成されることが示唆された. 一方,TtTPP91の立体構造および相互作用の解析を効率よく行うために,理化学研究所の平尾一郎博士と共同研究で,人工塩基対を利用した転写反応を行い,常磁性タグを特定の残基に導入した.この方法は,これまでに例のない画期的なものである.常磁性タグの存在によっていくつかのシグナルの強度が減少しており,常磁性タグの近くに位置していることがわかった.
I−8.J-motif RNA
J-motif RNAは,中村によって,神経系の分化に関連した遺伝子のmRNAに見出されたRNAの配列である.この配列は,vsfold/GPにおいては二次構造が予測されないが,実は構造を形成するという例であり,実際に特定の立体構造を形成しているかをNMR法によって解析することとした.塩基対に由来するシグナルが観測される領域において,J-motif RNAではシグナルが全く観測されなかった.しかしながら,重水中のNOESYスペクトルでは,3つのA残基のH2から非常に強いNOEが観測され,特徴的な構造の形成が示唆された.
I−9.長鎖RNAの相互作用の解明を行うためのQCMを用いた解析方法の開発
RNA−RNA,タンパク質相互作用を解析する新規な手法として,金センサー上にRNAを固定化することが可能な自己組織化膜を形成することで,ビオチン化などの煩雑な過程を必要としないQCM測定を実現するための基盤技術を開発することとした.本プロジェクトにおいて,種々の官能基を有するチオール誘導体を用いた,金基板上における自己組織化膜が形成されたQCMセンサーの調製に成功し,サケ白子由来のDNAを用いた固定化技術を開発するに至った.特に,末端にアミノ基およびカルボキシル基を有するチオール誘導体により修飾されたQCMセンサーを用いることで,効率的にDNAが固定化されることを見出した.さらに,坂本によって構造および相互作用が明らかとされているアプタマーであるApt1-Sを用いた実験の結果,アミノ基を末端に有するチオール誘導体により修飾されたQCMセンサーを用いることで,RNAが固定化されることを見出した.一方,酸化チタンを用いた光照射による生体分子の吸脱着制御についても開発を進めた.表面修飾を行った酸化チタンを用いることで,酸化チタンの光誘起超親水化現象に基づいた,光によるBSAの吸脱着制御に成功した.また,光照射による表面の濡れ性の制御が可能な薄膜の調製を行い,光照射により回収することが可能な繊維芽細胞のシートを調製するに至った.
チームIIでは,HIV-1およびHCVを対象として,その機能構造領域を見出すこと,および機能構造領域に対するアプタマーを取得し,それを機能制御に応用することをめざした.
II−1.HIVゲノムRNAにおける構造領域の機能の推定
(1)T細胞内miRNAによるHIV-1の複製制御
データベースに登録されているmiRNAと相補性を示すHIV-1の部分配列を含むベクターを,T細胞由来でHIV-1感染性のM4C8細胞とJurkat E6 cellsに形質導入し,抑制的な影響を持つ配列を多数同定した.その結果,pol領域,env-nef領域において新たな機能領域が示唆された.また,抑制効果を解除するように変異を導入したウイルスでは,ウイルスの産生量が減少する結果が得られた.これらの結果から,miRNAの抑制配列がウイルスゲノムのパッケージングとウイルスタンパク質の発現効率のバランスを担っている可能性が示唆され,HIV-1ウイルスがRNA 干渉に適応し,これを利用している可能性が示唆された.miRNAによる抑制効果を解除するようにpol領域およびenv-nef領域に部位特異的変異を導入したウイルスによる抑制の解除の確認,およびDrosha のknock-down およびRNA-IPによるAgpo2と標的mRNAとの相互作用の確認からも,本研究で見出されたmiRNAはHIV-1を標的とした宿主内miRNAである事が確認された.
(2)HEXM1-7SK snRNA-P-TEFb複合体は HIV-1 Tatの活性を制御する
HEXIM1は最近転写伸長反応の阻害装置として注目されている.本研究ではTat, HEXIM1とHIV-1 TARおよび7SK snRNAとの相互作用を検討することで,新たなLTR転写活性制御機構の解明を目的とした.TatおよびHEXIM1とHIV-1 TARおよび7SK snRNAとの相互作用をin vitroの系で解析したところ,HEXIM1と7SK snRNAの間には相互作用が無く,7SK snRNAはHIV-LTR転写活性には直接影響しないことが明らかになった.さらに,7SK snRNPがHIVの遺伝子転写の抑制には関わっていないことも示唆された.したがって,HEXIM1が過剰発現することでTARに直接結合してTatの機能を阻害したと考えられる.
II−2.宿主内因子,prostaglandin A1(PGA1)によるHCV-IRES依存的翻訳阻害
prostaglandin A1(PGA1)は,生体内のいたるところに存在して生理活性を有する物質であり,HIV-1などのウイルス感染時においては宿主防御因子APOBEC3Gの発現を高めHIV-1の増殖・複製を制御することを高久のグループが報告している.そこで,C型肝炎ウイルス複製制御に係わるPGA1の機能解析を行った.PGA1は,濃度依存的にHCVの複製を制御し,細胞毒性も伴うこともなく高い抗HCV活性を長期間示すことが確認された.また,PGA1はCap依存的翻訳阻害を伴うことなくHCV-IRES依存的翻訳阻害を誘導していることが分かった.さらに,PGA1は,PGA1/eIF3/40Ssubunit/IRES複合体を形成することでHCV-IRES依存的翻訳を阻害することが示唆された.
II-3.HIV ゲノム RNA における機能構造領域の応用
ウイルス遺伝子の発現を制御する際に最も有効な標的部位を探索することを目的として, DIS 領域に加え,ウイルス遺伝子のスプライシングに関与する4 つの部位(D1,D4,A3,A7)を標的とした核酸分子によるウイルス遺伝子の発現抑制効果を検討した.各部位を標的としたsiRNAを用いてレポーター遺伝子の発現抑制効果を比較した結果, DIS または D1 を標的としたsiRNAは,いずれもgag 遺伝子を含む mRNA の発現量を減少させた.またスプライシングによって生じるtat遺伝子の発現解析から,DIS または D1 を標的としたsiRNAがスプライシングを阻害している可能性が示唆された.つぎに,強い抗ウイルス活性が確認されたDISを標的としたshRNAについて発現ベクターを構築し,抗ウイルス活性を検討した.その結果,このベクターはHIV-1の遺伝子発現を抑制した.そこで,坂本が作製したTAR領域に対するアプタマーの抗ウイルス活性を同様な方法で検討したが,抗ウイルス活性は認められなかった. HIV-1遺伝子発現を制御する領域については絞り込みが出来たので,アプタマー不活性の原因を解明し,抗ウイルス活性を最大限に増強させることができれば,HIV-1治療戦略の選択肢を拡張するツールとして期待できると考える.
II−4.HCVゲノムの解析およびアプタマーの取得
HCVのタンパク質の一つであるNS5Aを用いてSELEXを行ったが,アプタマーを得ることはできなかった.一方,NS5AがHCVゲノムの3′-UTRに結合することを確認した.さらに,3′-UTR の欠損変異体を用いて相互作用解析を行ったところ,3′ X regionのSL IおよびSL IIを削った変異体および3′ X regionのみの変異体でもNS5Aと結合するが,3′ X regionのSLIを削った変異体では,NS5Aとの親和性が低くなることが示唆された.
II−5.HIVゲノムの解析およびアプタマーの取得
SELEX法とNMR法を用いて,HIVゲノムの5′-UTRの立体構造情報を得ることを試みた.アプタマーは標的分子の立体構造を認識して結合するので,得られるアプタマーの構造を解析することにより,標的分子の構造情報が得られると考えた.アプタマーは, 5′-UTRの分子表面と塩基対形成によって相互作用すると考えられ,塩基対を形成した領域のトポロジーから5′-UTRの全体構造をモデリングできる可能性がある.これまでSELEX法を立体構造解析に利用した報告はなく,新しい試みであると考えている.
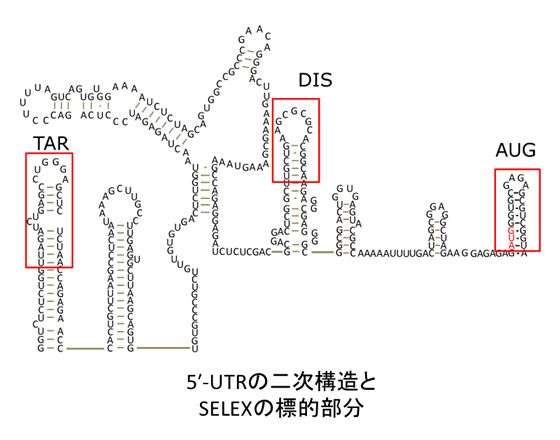
TARを標的としてSELEXを行なった.得られたアプタマー候補の配列を解析したところ,既に報告されているアプタマー配列を含むRNAの他に.それとは異なる配列も見つかった.新しいアプタマー候補のうち2つは,TARと相補的な配列を2箇所含んでいた.DISを標的としたSELEXでは,DISのループ部分に相補的な配列を含むライブラリと含まないライブラリを用いて選別を行なった.その結果,相補配列を含まないライブラリからもDISのループ部分に相補的な配列が濃縮された.一方,相補配列を含むライブラリからも同様な配列が濃縮されたことから,アプタマー配列の濃縮に成功していると判断した.開始コドンAUGを含むヘアピン部分を標的としたSELEXでは,標的RNAと結合するRNAプールが得られたが,配列の収束度が高くなかった.以上のように,5′-UTR に対するアプタマーが取得可能であることが示された(図).そこで,357残基の5′-UTR(SummersらのグループがNMR解析した配列)を調製し,樹脂に固定化して,SELEX実験を進めている.
5′-UTRのNMR解析も行っている.5′-UTRは,低イオン濃度(LI Buffer)ではモノマーとなるが,生理的条件(PI Buffer)ではダイマーになることが知られているが,NMR法によって,LI BufferとPI Bufferでは構造変化が起きていることを確認した.
II−6.長鎖RNAのX線結晶構造解析
HIV-1のゲノムRNAの5′-UTRは,立体構造が変化することによって,その下流にある遺伝子の翻訳を促す状態と,ウイルスゲノムがパッケージングされる状態とが制御されると報告されている.パッケージングの状態では,5′-UTRのDimerization Initiation site (DIS)を介して二量体化するとされており,この状態の立体構造を明らかにできれば,HIV-1粒子形成阻害剤の開発につながると期待される.そこで,HIV-1の5′-UTRの立体構造をX線結晶構造解析によって明らかにすることを目的とした.
HIV-1の5′-UTRの357merを T7 RNA ポリメラーゼを用いたin vitro 転写法によって調製し,600種類以上の条件で結晶化を開始した.目的とするRNA結晶は得られていないが,Mg2+存在下,あるいはTatタンパク質やヌクレオキャプシドタンパク質存在下において結晶化を試みる予定である.
以上のように,2つのチームのそれぞれにおいて成果が得られている.本プロジェクトにおいて,vsfold/GPによる長鎖RNAの二次構造の予測手法が開発でき,広くWebで利用可能となっている.また,RNAについてのNMR解析およびX線結晶構造解析の設備が整えられた.さらに,相互作用解析のための新しい手法も開発され,また,遺伝子発現制御の解析やアプタマーの取得などの研究もそれぞれ大きく進展した.これらの研究を推進するための装置・設備は本学津田沼キャンパスに集約されており,まさに研究拠点が形成されたと考えている.以上のことから,本プロジェクトは当初の目標を超える成果を上げたと考えている.